
Testing frontier AI models for a fast follower campaign: from target selection to chemical hypotheses
Why the most useful answer was no

Science is not just about answers, it’s about testing the feasibility of those answers. In science, negative data is as or more important than positive data because it can prevent you from going down blind alleys and wasting resources. When I ask an AI a question, I don’t want it to answer the question no matter what. I want it to tell me if an answer is even feasible. The entire workflow took about two hours.
I wanted to test a harder question than whether an AI-assisted workflow could dock compounds or build an ADME model; we are already there now, and I have presented examples in the past. Instead, could it look across an entire target family, weigh the biology, chemistry, clinical evidence and commercial landscape, and then tell me that I should not start the program I had asked it to start?
Cyclin-dependent kinases are a good test case. The family contains some of the most clinically validated kinase targets in oncology, several crowded emerging classes, and a long tail of understudied CDKs that look attractive precisely because less is known about them. This creates a familiar drug-discovery trap. The well-validated targets may have little room left, while the apparently novel targets may be “open” because the biology, therapeutic index or chemical tools are not ready.
The goal was therefore straightforward to state but hard to execute: survey the entire CDK family, find the best differentiated fast-follower opportunity, and then use public chemistry and structures to propose selective chemical starting points.
The exact scientific request was:
I want to find a novel CDK and novel leads against it. Perform a survey of existing CDKs and their chemical matter and identify the best one for a fast follower campaign. Build and use a statistical validation agent for doing the modeling based on multiple scientific and business axes necessary for picking the target. Register this agent in the Bio Agent fleet for reuse.
Then do a structure-based design campaign for that CDK by starting from a
roughly pan-CDK library and identifying selective inhibitors.
Also consider non-CDK inhibitors based on 2D and 3D similarity from other targets
like GPCRs and nuclear receptors. Once the top selective hits are prioritized,
build ADME models for solubility, permeability and metabolic stability using
public data to further prioritize.
Work autonomously, but before you execute this campaign, list the plan along
with agents, tools and capabilities you will be using.I added two implementation constraints around the preferred chemistry stack and tool transparency. More on those details at the end; the science is the more interesting part.
The first screen was not a virtual screen
It was tempting to begin with a structure and a thousand compounds. That would also have been the wrong first step.
Before spending computational effort on molecules, the campaign treated target choice itself as the primary model. All 20 CDKs were evaluated across 14 axes spanning human biology, disease and biomarker logic, therapeutic window, mechanistic differentiation, structural tractability, public chemical matter, assay readiness, clinical precedent, competitive crowding, market whitespace and speed to a defensible development candidate.
The key methodological point was that weighted scores were not allowed to overrule fatal criteria. A target could have excellent chemistry and still be a bad program if it had reasonably validated on-target toxicity. A target could also have exciting biology and still be too early to call a fast follower.
The predeclared gates asked six basic questions:
Is there a defensible indication?
Is there a plausible therapeutic window?
Is there a credible route to target and isoform selectivity?
Are the target state, assays and structures fit for purpose?
Is there enough real starting matter rather than database noise?
Is there a differentiated target product profile relative to incumbents?
The statistical layer then ran deterministic multi-criteria ranking, Pareto analysis, missing-data penalties, leave-one-axis and leave-one-evidence-family sensitivity, and 20,000 uncertainty simulations. These simulations measure sensitivity to assumptions. They are not probabilities of clinical success.

Figure 1. Relative target ranking and absolute gate eligibility are different questions. The weighted leader was not an eligible target, and no CDK passed every fatal gate.
The initial scale was:
20 CDK alternatives
14 scientific, chemical, clinical and business axes
20,000 value-and-weight sensitivity simulations
6 predeclared fatal gates
0 gate-eligible targetsThat last line was the most important result in the campaign.
The answer was no
As anyone who has done drug discovery knows, target selection and assessment are incredibly hard, in part because you can always nominate some target based on a few favorable criteria. Bad targets based on partial data are picked with good faith all the time. I had explicitly asked for the best CDK. A less skeptical workflow could easily have produced one. It could have sorted the weighted scores, selected the highest underexplored kinase, drawn a few plausible poses and called the exercise a success. Instead, the AI’s frank assessment was that no target really fit the bill.
This is exactly the behavior I want from an AI assistant doing science. An assistant that must always return a positive answer is a suggestion engine. A useful scientific assistant has to be capable of falsifying the premise of its own assignment. It also shows that the models are getting better, since I used an earlier model and got a confident answer that picked a specific target.
There was no single large, clean CDK signal. The mature targets were usually strong for the same reasons they were commercially crowded. The obscure targets were often uncrowded because they lacked human validation, a selective chemical probe, a credible biomarker or even a well-resolved assay strategy. “Whitespace” turned out to contain several different things, not all of them opportunity.
The family separated into a few recognizable groups:

This distinction matters economically as much as scientifically. A negative target recommendation can save far more value than squeezing another decimal place out of a docking score. Months of chemistry on the wrong target are still months of chemistry.
CDK11 was the most instructive near-miss
CDK11 initially looked like the sort of target a fast-follower exercise might favor. It had unusually good structural and chemical tractability relative to its clinical maturity. There was a plausible tumor-sensitivity hypothesis around 1p36 deletion, public tool matter and a visible route to structure-based optimization.
Then the biological decision gate failed. A resistance-allele study developed MEL-495R, a selective and orally bioavailable CDK11 inhibitor, and used an engineered mouse system to distinguish on-target from off-target toxicity. The study attributed widespread toxicity to intended CDK11 inhibition and found that tolerated exposures did not provide the efficacy window needed for a conventional systemic program. That is not a minor developability issue. It directly challenges the target-level therapeutic hypothesis. The primary paper is unusually valuable because it deconvolutes efficacy and toxicity genetically.
At the same time, the space was no longer commercially empty. Acrivon reported a CDK11 development candidate and backups in IND-enabling studies, with an IND for ACR-6840 or an alternative candidate planned for the first half of 2027 in its May 2026 SEC disclosure.
CDK11 therefore combined two problems: an incumbent and a target-level safety warning. Excellent chemistry could not rescue it. The systemic program was stopped.
This may be the most scientifically useful negative result in the whole exercise. The workflow did not confuse tractability with desirability.
CDK8/19 was the best conventional follower—and still not good enough
CDK8/19 came out as the strongest less-crowded clinical follower. It had meaningful human execution precedent and a more conventional fast-follower profile than the obscure CDKs. RVU120 is a selective dual CDK8/19 inhibitor with an active Phase 2 program in myeloid malignancies, including AML/MDS and myelofibrosis.
But “best available” was not the same as “good enough.” RVU120 already occupies the dual CDK8/19 space across myeloid malignancies, and the campaign did not identify a credible product profile that was clearly superior on selectivity, indication, biomarker, schedule or combination logic. CDK8/19 remained a benchmark, not a nomination.
The distinction is subtle but important. Ranking asks which alternative is best relative to the others. A development gate asks whether that alternative is good enough in absolute terms. Drug discovery needs both questions.
Why CDK12 was retained as a conditional chemistry benchmark
CDK12 was not declared the winner. It was retained because it offered the clearest experimentally testable differentiation hypothesis among the emerging targets.
CDK12 and its close paralog CDK13 are transcriptional kinases activated by Cyclin K. They regulate RNA polymerase II and the expression of long genes, including DNA-damage-response genes. The pharmacology is already rich but mechanistically entangled. THZ531 inhibits both CDK12 and CDK13. SR-4835 is a reversible CDK12/13 inhibitor. CT7439 is in a recruiting Phase 1/2 study, but Carrick describes it as a dual CDK12/13 inhibitor and Cyclin-K glue degrader, not a clean CDK12-selective inhibitor. The trial has no posted results, and the sponsor’s description makes the mixed mechanism explicit.
That left a narrow but interesting hypothesis:
Could a reversible, non-degrading inhibitor meaningfully spare CDK13 while inhibiting CDK12 in a biologically selected CDK12-intact or CDK12-upregulated tumor context?
Every word in that sentence matters. CDK12 loss can behave as tumor-suppressive loss, so “CDK12-mutant” cannot simply be copied from a genomic report and treated as an inhibitor biomarker. The intended campaign would have to distinguish selective CDK12 inhibition from dual CDK12/13 inhibition and from Cyclin-K degradation. It would also need to prove that the selected tumor state is dependent on CDK12 activity rather than already lacking it.
Those unresolved biomarker and therapeutic-window questions are why CDK12 failed the development gates. They are also why it was useful as a research benchmark: the selectivity hypothesis was precise enough to test.
The structure-based campaign
The receptor was the human CDK12/Cyclin-K complex bound to SR-4835, PDB 8P81. This is a 2.68 Å crystal structure and captures a non-canonical glycine-rich-loop conformation around the reversible inhibitor.
Before prospective screening, the crystallographic ligand was removed, regenerated and re-docked. The symmetry-corrected heavy-atom RMSD was 0.456 Å, comfortably below the predeclared 2.0 Å RMSD. That does not prove prospective enrichment, but it does show that the preparation and docking protocol could reproduce the crystallographic binding mode.
Two chemical branches were then prepared:
Pan-CDK ChEMBL-derived library
1,200 standardized molecules
45,133 conformers
GPCR and nuclear-receptor cross-target library
1,086 standardized molecules
43,159 conformers
Total prepared conformers
88,292The first branch asked a familiar medicinal-chemistry question: could chemical matter reported against other CDKs hop into CDK12 while gaining a panel selectivity signal?
The second branch was deliberately less conventional. It used 2D fingerprint similarity and 3D shape/color overlays to search GPCR and nuclear-receptor ligands for molecules that resembled the CDK12 crystallographic ligand in ways that were not obvious from target annotation alone. This was a scaffold-repurposing exercise, not a claim that GPCR or nuclear-receptor pharmacology predicts kinase inhibition.

Figure 2. The pan-CDK scaffold-hopping and cross-target 2D/3D similarity branches were filtered independently before converging on a 12-compound experimental set.
The pan-CDK branch produced a CDK12 pose for 1,199 of 1,200 compounds. Of these, 1,174 had sufficiently complete scores against the CDK anti-target panel, including CDK13. Percentile filtering retained 113 molecules, and fingerprint diversity reduced this to 25.
For the cross-target branch, all 1,086 compounds received 2D similarity scores. The top 300 3D overlays were retained, the union of 2D and 3D evidence produced 215 candidates, and 100 diverse molecules were docked against CDK1/2/4/5/6/7/8/9/12/13. Seventeen passed the CDK12, CDK13 and broader panel filters; 15 survived final diversity selection.
The scores were converted to within-receptor percentiles. This was important because raw docking scores from different receptor preparations are not calibrated affinity measurements. A positive CDK12-minus-CDK13 percentile difference is a prioritization signal, not biochemical fold selectivity.
What came out
The final set contained 12 compounds: pan-CDK scaffold hops, cross-target shape-repurposing hypotheses and two known CDK12/13 control-like comparators.
The highest-ranked compound was CHEMBL3698676, a scaffold-hop hypothesis with public CDK4/6 data. It had a CDK12 docking percentile of 0.989 and a CDK12-minus-CDK13 percentile delta of 0.720. Those numbers made it a good first test article. They did not make it a CDK12 inhibitor.
Other notable hypotheses included:
CHEMBL546797, a pan-CDK hop with public CDK7/8/9 data and a strong modeled CDK13-sparing signal, but with oxime/imine-related chemistry alerts.
CHEMBL2003456 and CHEMBL4225966, relatively clean-alert scaffold-hop hypotheses that still require counterscreens against their previously annotated CDKs.
XTARG_00994 and XTARG_00998, NR4A2 agonist-derived shape hypotheses that need explicit NR4A2 controls.
XTARG_00414, a CNR2 ligand with substantial lipophilicity and solubility risk.
CHEMBL4211454 and CHEMBL4868958, CDK12/13 control-like compounds included as comparators rather than novel matter.

Figure 3. OpenEye-depicted conditional test set containing pan-CDK scaffold hops, cross-target shape hypotheses and two control-like CDK12/13 compounds. The displayed values are docking percentiles, not measured affinity or fold selectivity.
Every final pose passed a permissive geometry check: no protein heavy-atom clash below 1.2 Å and at least two contacts shared with the crystallographic pocket. Since all 12 passed, this gate established geometric plausibility but provided essentially no ranking power. That itself is worth stating. A quality-control metric that every candidate passes should not be advertised as independent validation.
The compounds were therefore labeled with deliberately narrow language:
predicted CDK12-binding hypothesis
predicted CDK13-sparing hypothesis
experimental test articleNot “hit.” Not “selective inhibitor.” Certainly not “lead.”
The ADME models also knew when to say no
The campaign built public-data models for aqueous solubility, Caco-2 permeability, microsomal clearance and hepatocyte clearance using endpoint definitions from the Therapeutics Data Commons. The solubility and permeability models had respectable held-out performance under scaffold-grouped splits:
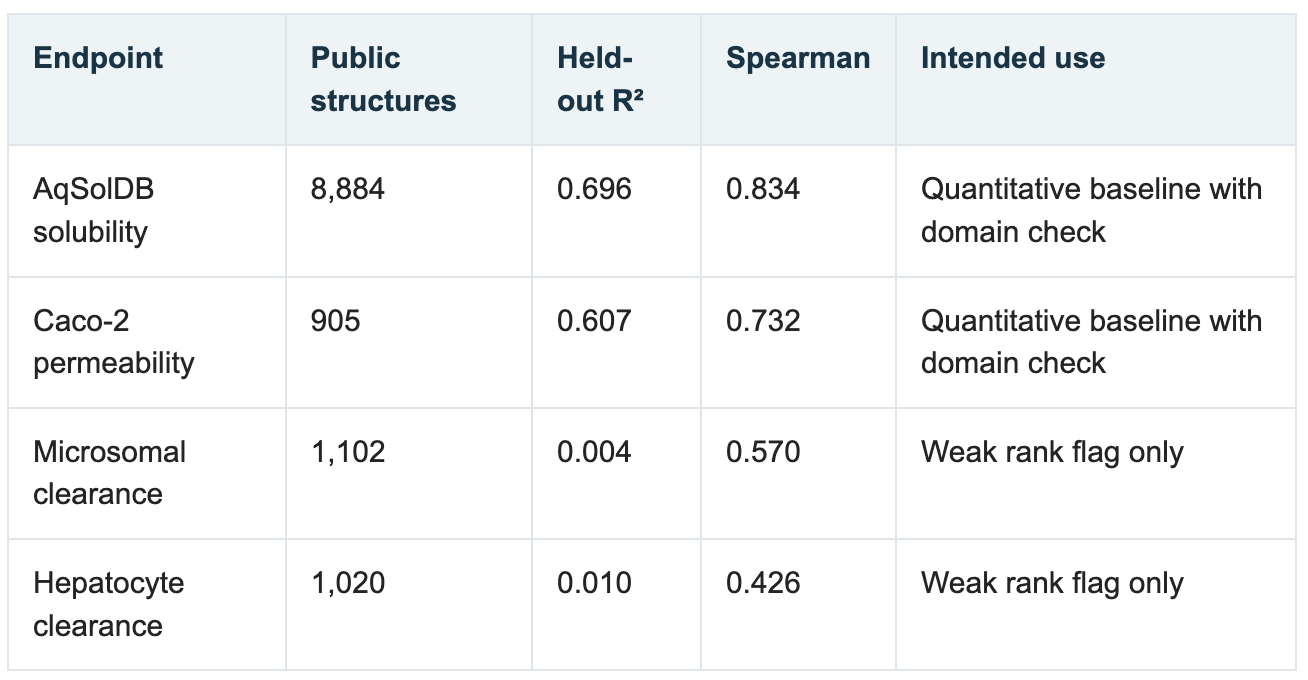
The clearance models were plainly weak. More importantly, nearest-neighbor applicability analysis showed that the candidate chemistry sat far from much of the public training data. Among the final 12 compounds, 11 were classified as extrapolative, no decision. Only CHEMBL546797 reached a borderline baseline domain.
This is another example of a negative result being scientifically useful. The models did not become reliable merely because they had been trained successfully. The applicability analysis prevented a technically valid model from turning into an invalid project decision.

Figure 4. Solubility and permeability models had useful held-out performance, but applicability—not nominal model quality—was the decisive limitation for the proposed chemistry.
The practical result was that the models could order assays and flag obvious risks, but they could not replace measured solubility, permeability or metabolic stability.
What I would test next
The 12 compounds are best treated as a blinded hypothesis set. The first experiments should be designed to eliminate false positives and resolve the central mechanism, not to beautify a computational ranking.
I would run them in this order:
Identity, purity, measured solubility, aggregation and optical-interference controls, with reactivity testing for flagged chemotypes.
Matched CDK12/Cyclin-K and CDK13/Cyclin-K biochemical assays in the same format and ATP conditions, plus an orthogonal binding assay.
Cellular target engagement for both kinases and time courses for Cyclin K, CDK12 and CDK13 abundance.
CDK1/2/4/5/6/7/8/9 counterscreens, followed by a broader kinome panel for confirmed actives.
NR4A2, CNR2 and AVPR1A counterscreens for the cross-target hypotheses.
Measured solubility, Caco-2 or MDCK permeability, microsomal stability and hepatocyte stability.
Comparison of selective-CDK12 and matched dual-CDK12/13 pharmacology in held-out CDK12-intact or CDK12-upregulated models, together with normal-cell transcriptional, marrow and gastrointestinal safety systems.
Only a compound that confirms CDK12 potency, meaningful CDK13 sparing, absence of Cyclin-K degradation, clean counterscreens and measured developability should graduate from “test article” to “lead.”
Why the skeptical layer matters
There is a natural concern that AI-assisted research will be biased toward fluent positive stories. The model is asked to find a target, so it finds one. It is asked to find hits, so it returns hits. It is asked to build an ADME model, so it produces predictions with three decimal places.
This campaign is a useful counterexample.
The target-ranking model said no CDK passed the gates. The independent skeptic challenged the apparent winners. CDK11 was stopped despite attractive chemistry. CDK8/19 was not advanced merely because it was the best conventional follower. CDK12 was explicitly demoted from “recommended target” to “conditional benchmark.” The docking results were called hypotheses rather than affinity. The pose-contact gate was identified as non-discriminatory. The ADME models were prevented from making decisions outside their applicability domain.
In other words, the negative conclusions were not failures of the workflow. They were the scientific product.
The best computational campaign is not necessarily the one that produces the longest list of compounds. Sometimes it is the one that makes the list smaller. Sometimes it redirects the question. Sometimes it says that the evidence is not yet good enough and tells you exactly what experiment would change the answer.
That kind of skepticism is valuable because drug discovery already contains more than enough ways to rationalize a positive result after the fact.
The computational implementation
GPT 5.6 Sol implemented in Codex was used for all investigations. A Bio Fleet agentic stack was manually created based on well-known public tools like ChEMBL and PubChem. Instructions were vibe coded in the agents.md and skill files. The campaign used separate scientific/structural, chemistry, clinical/market, statistical and skeptical roles. Orchestrator, Skeptic and Integrator agents were defined by Codex. Their outputs were written as independent claim bundles with evidence, confidence, risks and recommended actions.
The reusable statistical component, cdk-opportunity-statistics, was packaged and registered in the Bio Agent fleet. It performs schema and direction checks, gated multi-criteria analysis, uncertainty propagation, Pareto analysis, missingness stress tests, evidence-family sensitivity and data-split leakage audits. The point of making it reusable is that the same skeptical machinery can be applied to other target families.
Codex coordinated the campaign, maintained the artifact and provenance registers, ran the chemistry and modeling workflows, and assembled the final interactive HTML report. The report retains the exact prompt, target evidence table, statistical sensitivities, receptor validation, screening funnel, all 12 structures, pose cautions, ADME model cards, experimental gates and links to machine-readable outputs.
The two chemistry-specific follow-up instructions were intentionally brief:
One tweak: wherever you can use OEChem instead of RDKit use it. But use your best judgment.
Also list what OpenEye tools you intend to use.For the chemistry, I used OpenEye tools wherever they were a good fit. OpenEye tools were installed and run purely from the prompt. I ran in a mode that asked Codex to use OpenEye tools whenever applicable and others when needed. The following tools were automatically and autonomously invoked without explicit calls:
OEChem for structure parsing, standardization, identity handling and molecular I/O.
OEMolProp for molecular descriptors.
OEGraphSim for fingerprints, diversity, 2D similarity and applicability-domain calculations.
OEOmega for multiconformer generation.
OEQuacPac for protonation and charge preparation.
OESpruce for receptor preparation and design units.
OEDocking for redocking and CDK-panel pose generation.
OEShape/ROCS for 3D shape and color overlays in the cross-target branch.
OEDepict for the final compound structure package.
RDKit was retained for two independent checks where it added value: Murcko scaffold grouping for model splits and a secondary catalog of PAINS, Brenk and NIH structural alerts. ChEMBL, BindingDB and PubMed were used for literature search and data extraction.
The full run produced a reviewable, reproducible campaign rather than a folder of disconnected calculations. That distinction matters. The value was not any single algorithm; it was the connection between target biology, commercial context, chemical evidence, structural validation, model applicability and explicit experimental decisions.
Closing thought
I began by asking for a novel CDK and novel leads. The rigorous answer was that no CDK currently justified a development nomination, and none of the proposed compounds could yet be called a lead.
That may sound less exciting than a list of “AI-discovered inhibitors.” I think it is much more useful. A positive computational hypothesis gives you something to test. A well-supported negative conclusion tells you what not to spend the next year doing. In drug discovery, both are progress.
Acknowledgment: Many thanks to OpenEye Scientific for providing an evaluation license for this project. The license made the OpenEye-first chemistry, docking, shape-similarity and depiction workflows possible. The scientific conclusions and any errors are my own.
Scientific note: This was a computational hypothesis-generation exercise with an evidence cutoff of July 9, 2026. It is not a patent/FTO, clinical, regulatory or investment opinion. No compound should be considered a confirmed CDK12 inhibitor or lead without experimental validation. The opinions offered in this analysis are mine alone and do not reflect those of my employer or any other entity.