
Running an AI-assisted computational drug repurposing project over a weekend
Computational chemistry at warp drive speed

I wanted to see whether Codex could run a serious computational drug-repurposing workflow, end to end, if I treated it less like a chatbot and more like a scientific computing colleague with a well-defined lab notebook. To do this I ran an entire multi-stage computational drug repurposing campaign using Codex (Pro tier) autonomously on my MacBook Pro M5 laptop over a single weekend. My goal in the following post is purely educational and not to single out Codex; I just choose to use it because of familiarity and budget reasons. It’s rather to make the point that we are now in a golden age in which most frontier AI models and AI automation can accomplish what I accomplished with Codex. What I want to drive home is that if you are a computational scientist working in drug discovery or biotech, being equipped with these tools is no longer a choice; it’s a necessity. More importantly, it’s a tremendous opportunity.
The test case was IL-17A, an old target that I worked on at my first job. IL-17A is an important inflammation and immunology target implicated in psoriasis. Inhibiting it is hard because it forms a PPI.
The goal was straightforward and typical but nontrivial: find repurposing candidates with a docking-based selectivity signal for IL-17A over related IL-17 family members, then carry the hits through property filtering, patent triage, short MD, solubility modeling, and a final scientific report.
The actual scientific prompt was intentionally high-level, because the whole point was to have the LLM abstract away the tools, databases and models:
I want to discover new selective IL-17A inhibitors.
They should have at least a 3 to 5 fold selectivity over other IL-17s.
Start by docking a diverse set of at least 1000 pan IL-17 inhibitors and drugs
against IL-17A and other relevant IL-17s.
Exclude already known high affinity IL-17A binders except a few positive controls.
Generate and prioritize the top 100 structures using docking scores and
filter using physicochemical properties.
Then run a patent search on these 100 to identify novel compounds.
Run short MD simulations on the top compounds for stability assessment.
Finally, build a predictive ADME model for solubility and prioritize
the most soluble compounds.
Act autonomously. Infer tools, retrieve data, install missing dependencies
if necessary, create reproducible outputs including a Jupyter notebook
and document all assumptions and reasoning.
As anyone who has run computational campaigns from scratch knows, this kind of multistep, multi-tool workflow can easily take days if not weeks to set up and run and debug. Instead it took me a little more than two days, but almost the entire 48 hours from that time was just the system running autonomously without any human input; my own work effectively consisted of going from installing Codex to configuring and launching this workflow and analyzing the results in a total of about three hours.
I would say the most important part of the setup was not the prompt. It was the AGENTS.md file. I want to emphasize that the name is something the system picked: this is not an “agentic” workflow. The name is thrown around a lot these days, but this workflow just depended on the capabilities of GPT 5.5. Real agentic workflows can get much more powerful.
Think of AGENTS.md as the master controller for the project. It’s kind of a superpower, in fact, since subtle changes in this file can significantly impact the shape and nature of your results. This is where you tell Codex how you want science done in your environment: which tools to prefer, how autonomous it should be, what outputs to save, what assumptions to document, and what level of evidence is acceptable. You can say pretty much anything you want in this file, and it’s fascinating to experiment with it.
A typical AGENTS.md file would start like this:

That file changed the character of the run. Instead of asking for one-off commands, I was giving Codex an operating style.
The file also contains a list of individual tools with instructions on how to use them. You can either manually ask the file to include these tools or ask the LLM to infer them, and the system will just populate the file with all relevant details. For instance for RDKit and Autodock Vina this is what the LLM populated the AGENTS.md file with:

You can of course tweak every parameter in this description. But here’s the interesting thing: I never specified RDKit or Autodock Vina as the tools. I simply said in the AGENTS.md file, “The user should be able to describe the scientific goal without naming the software. Infer and install the right tool, choose reasonable defaults, run the workflow, and summarize results.” For an average scientist this is a huge value add, since you often don’t know beforehand what tool you might need to execute a workflow. In fact, inferring tools, databases or models from intent alone is the essence of what a scientist wants.
I also added a small HTML report skill.md file. Its job was simple: when the calculations were done, assemble the evidence into a report that a computational chemist could actually review and present. Tables, molecule grids, docking plots, selectivity plots, MD traces, caveats, data provenance, and links back to the generated files. All the details that you might want for a presentation reside in this report.
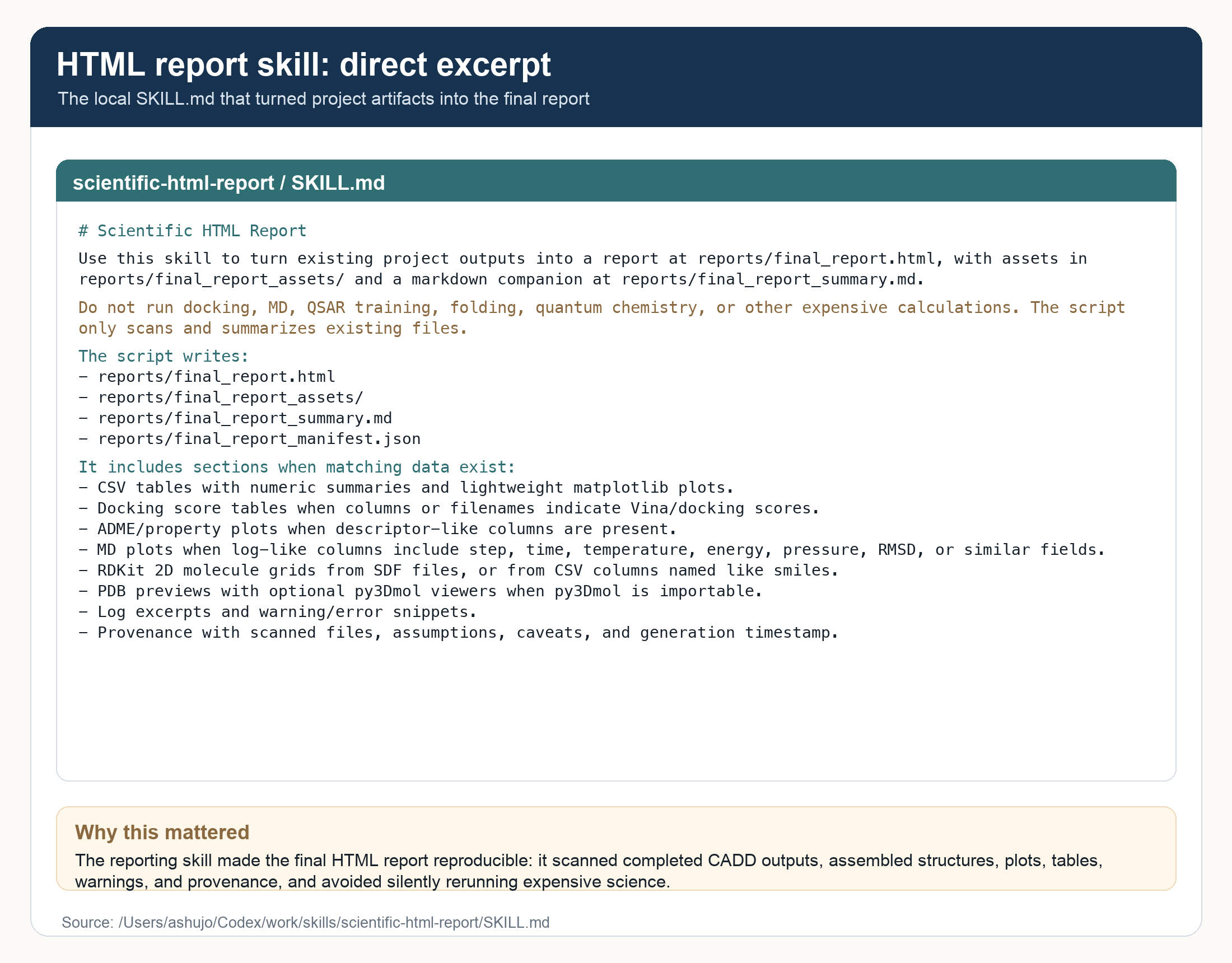
What came back after 48 hours was a full local CADD campaign. The workflow collected public IL-17-related activity records and approved drug-like compounds from ChEMBL and Pubchem, standardized the structures with RDKit, prepared receptors for IL-17A, IL-17F, IL-17A/F, and IL-17E, and ran a high-exhaustiveness Vina screen across the receptor set. All without a single manual input.
Scale:
1001 ligands
3984 planned docking tasks
3910 completed dockings
Top 100 retained for review
3 explicit-solvent OpenMM MD checks
47.99 hours of continuous wall-clock execution
That 48-hour stability is the part I still find impressive (it was also a good stress-test for my laptop!). It means Codex was maintaining a project state across public-data retrieval, receptor prep, ligand prep, docking, failed-job handling, ranking, patent triage, solubility modeling, MD setup, MD execution, analysis, figure generation, and report writing for that long.
Here’s the full table detailing the task, tool and corresponding wall clock time that shows the scope of what ran autonomously.
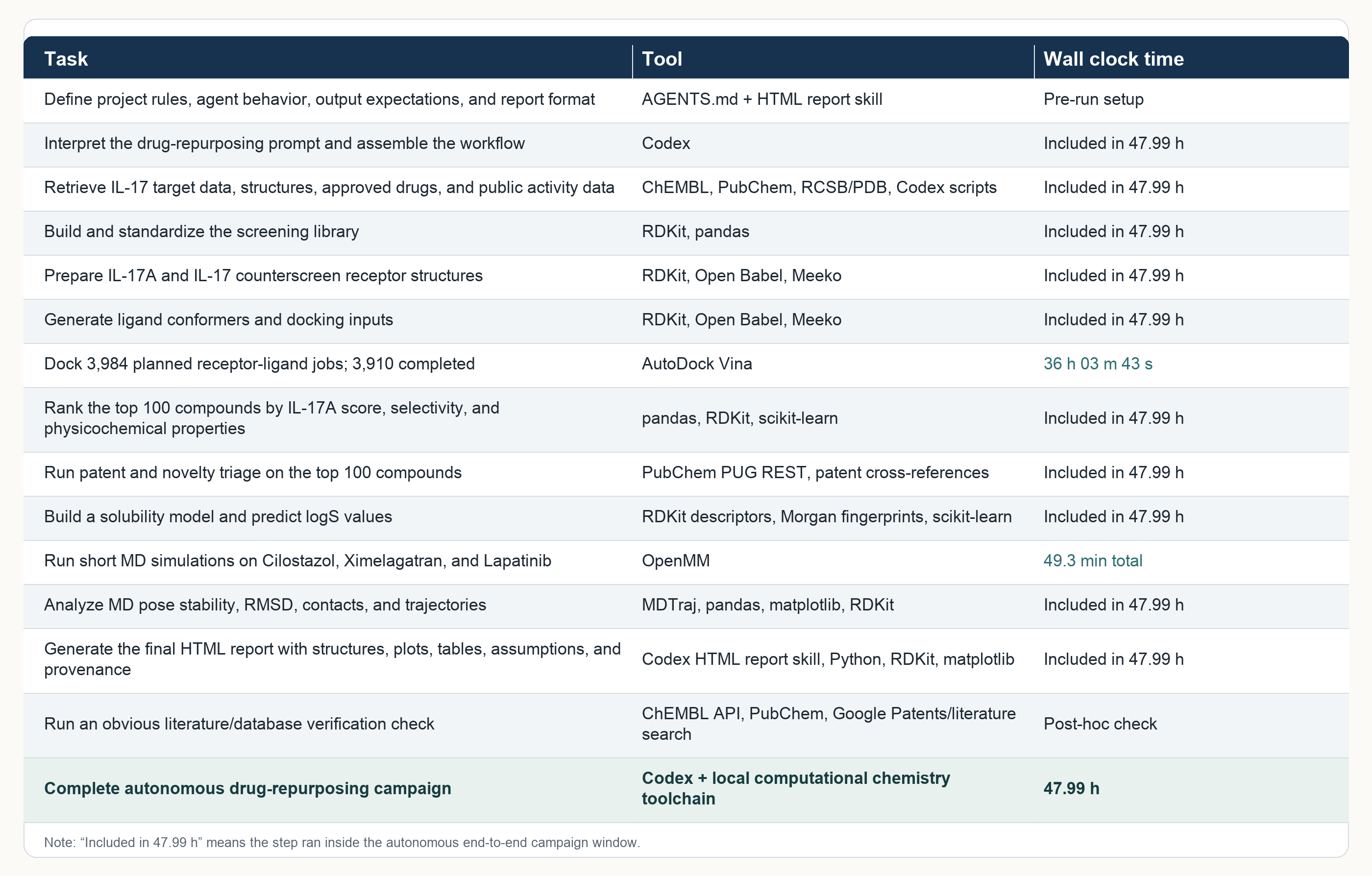
Anyone who has run these workflows by hand knows that the hard part is often the connective tissue: file formats, missing dependencies, failed ligands, stale paths, half-written logs, tables that no longer match plots, and the quiet accumulation of tiny mistakes. Having a workflow remain coherent across that whole span changes what a single scientist can attempt in a weekend.
The HTML reported included a variety of structure and property-based plots.
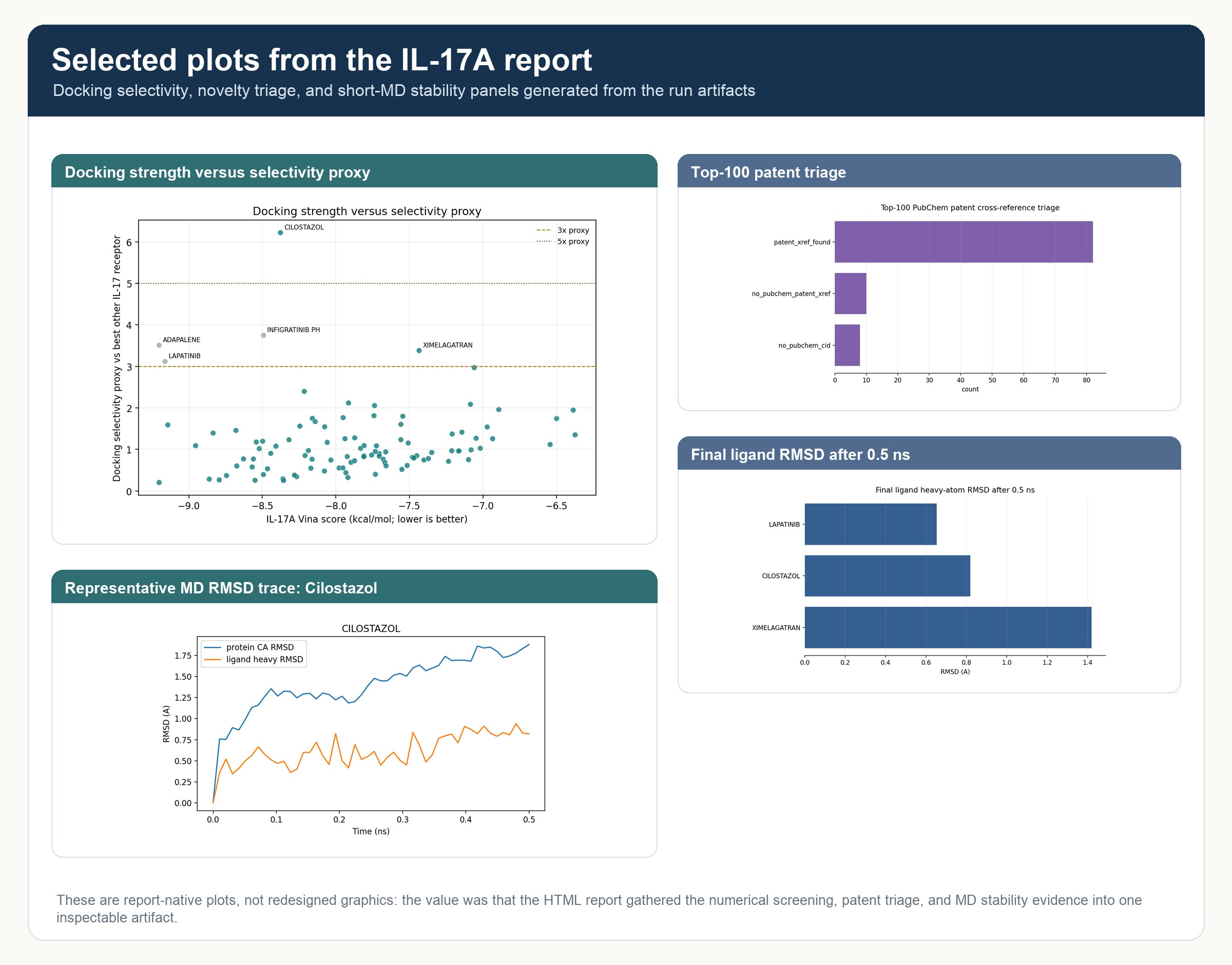
The top computational hypotheses were Cilostazol, Ximelagatran, and Lapatinib. I have no idea whether these drugs could be repurposed for IL-17 (someone should try!); in fact Ximelagatran probably would be a non-starter because it was withdrawn because of liver toxicity. But the point is that all three structures came from a scientifically plausible chain of computational reasoning, of the kind that drug discovery scientists build all the time.
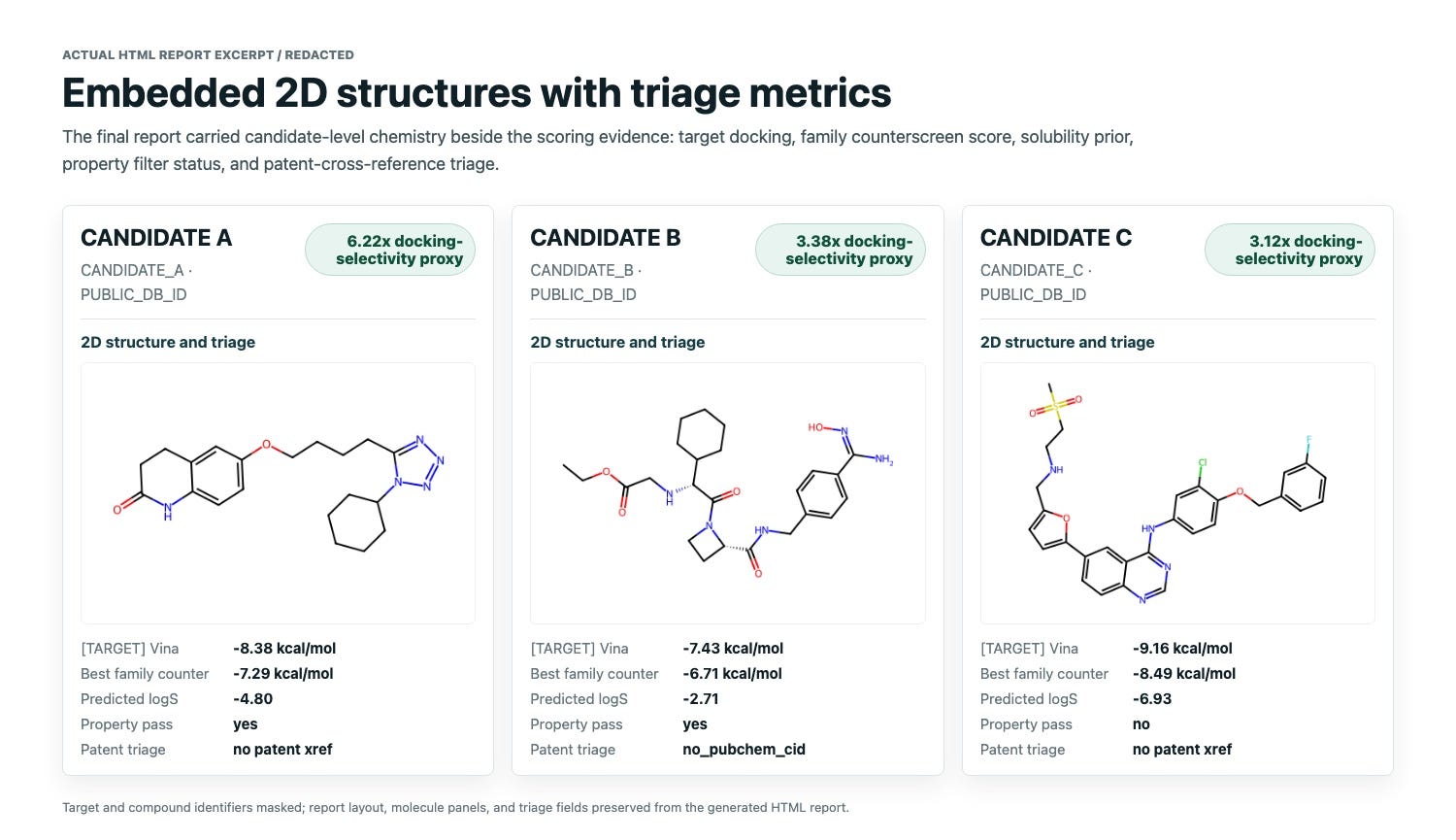
Just as importantly, the report did not overclaim. It explicitly treated docking and short MD as triage, and it did flag small differences in docking scores which may not be significant. Before writing this up, I also did the obvious sanity check: were these already listed as IL-17A inhibitors so that the entire workflow was just a lookup? Public ChEMBL target-activity checks did not show direct IL-17A activity records for these three compounds. Lapatinib had the most important prior signal: literature and patent evidence connects it to upstream Th17/RORgamma biology and reduced IL-17A secretion. That is useful context, but it is different from direct IL-17A binding or IL-17A/IL-17RA blockade. The fact is that Cilostazol, Ximelagatran, and Lapatinib were not chosen by the system because of known IL-17A activity. They entered the screen as part of a broad ChEMBL max-phase drug repurposing library, then surfaced after a rational process of docking, selectivity, property, solubility, patent, and MD triage.
The final HTML report was a good payoff. It was not just a file dump. It was a reviewable scientific artifact: executive summary, original prompt, interpreted plan, receptor setup, docking sanity checks, compute/runtime summary, ranked candidates, property filters, patent triage, solubility model, 2D structures, MD complex images, RMSD traces, caveats, recommendations, and data provenance. Exactly the one-stop shop you need for summarizing the investigation.
For me, the lesson is practical:
If you want Codex (or any other similar system) to do useful computational drug design work, do three things.
Create an AGENTS.md file that defines your scientific standards, preferred tools, autonomy level, file conventions, and reporting expectations.
Give it access to a real project folder with the right local environment: RDKit, Open Babel, Vina, OpenMM, scikit-learn, pandas, plotting tools, and enough write access to create reproducible outputs. It will use or install all these tools as needed. One thing to watch out for is the level of access you want to give Codex. You could technically give Codex access to your entire computer, but OpenAI themselves caution against this for good reasons. Having a sandboxed environment is a good starting point.
Ask for a scientific objective, not a list of shell scripts. Then require the final answer to be a report that exposes assumptions, failures, caveats, and provenance.
This does not remove the need for experimental validation or medicinal chemistry judgment, of course. Those become even more important for verification and further inquiry. But it does change the amount of computational triage one person can do. The fact is that in a few hours of setup, followed by a stable two-day autonomous run, Codex produced a repurposing evidence package that I could inspect like a real CADD report, one that would have taken me days or weeks to produce otherwise.
If this set of experiment-ready hypotheses is what I could create over a weekend, imagine what a computational chemist could create in a week, a month, a year. The sky is the limit. New ideas for experimental testing would pour out from these workflows like water, with only the human need for sleep and compute limiting them. Scientists who work on two or three projects could now work on ten. Human oversight would of course be necessary, and how to provide this oversight as the projects scale is its own question.
But the bigger point should not be lost here. AI can now easily do the kind of not just routine work but complex pipelining that computational scientists do, and it can do this much faster and at higher scales. It’s hard to escape the simple conclusion: AI-assisted computational drug design is the future. If you aren’t on that train you are losing out big time, and you may well be left behind. I don’t see this as a threat or a warning. In fact I see it as an immense opportunity, possibly the biggest one we have seen in a very long time.