


Classics in Medicinal Chemistry #1: Atorvastatin
Lipitor


There’s no better candidate for a medicinal chemistry historical chronicle than atorvastatin (Lipitor), the best-selling drug in history. Approved by the FDA in 1996, this small molecule revolutionized the treatment of hypercholesterolemia and the prevention of cardiovascular diseases, which remain among the leading causes of morbidity and mortality worldwide. Over its lifetime, the drug has generated an astonishing $150 billion globally. Even now, after the patent has expired, it generates around $2 billion. The success of Lipitor appears even more striking when you realize that it was the fourth statin on the scene, marketed ten years after the first statins appeared.
Atorvastatin’s stunning success was in part due to Pfizer’s clever marketing and in part due to atorvastatin’s superior pharmacological properties. But as medicinal chemists and drug discovery scientists, it is the science behind atorvastatin’s design that interests us the most. Many publications describe the synthesis, properties and testing of atorvastatin analogs, but a convenient publication that gathers all the information together is from the lead inventor, Bruce Roth, and that is what we will be looking at today (unless indicated otherwise, all images are from this paper).
The origins of the statins lie in the discovery of fungal metabolites from the 1970s and 80s that led to the first statin drugs - compactin and lovastatin. Statins work by inhibiting HMG-CoA reductase, a key enzyme in cholesterol biosynthesis, effectively lowering LDL cholesterol levels in patients.

Image: Position of HMG-CoA in the cholesterol biosynthesis pathway (Image Credit: Frontiers in Oncology)
Most of the cholesterol we have in our body (about 60%) is biosynthesized, so even controlling our diets limits how much we can impact hyperlipidemia and its resulting complications like atherosclerosis.
The discovery of atorvastatin was a testament to many important aspects of medicinal chemistry and drug design:
The application of basic molecular modeling and 2D comparisons.
The use of classical (19th century!) heterocyclic chemistry and the development of a chiral synthesis.
The close collaboration between discovery and development in optimizing the synthesis and scale-up.
Some tough decision-making by Pfizer leadership in advancing the drug into clinical trials, even in the face of unfavorable biological readouts.
In our age of sophisticated chemistry and CADD techniques, it is refreshing to see how (relatively) simple tools could result in the invention of such an important drug.
The publication starts by noting that although four other statins were discovered in the 1980s, safety concerns led to temporary suspensions in the development of drugs like lovastatin. Thus there was a perceived need for structurally novel inhibitors of HMG-CoA reductase that would not have these liabilities while preserving potency and PK/PD properties.
If we look at the structures of the previous drugs, it is clear that they share a common hexahydronaphthalene core connected to a mevalonolactone moiety: effectively a part that mimics the transition state of the reaction catalyzed by HMG-CoA reductase connected to a large, lipophilic part. The logical question was: could you replace the lipophilic part with other ring systems? The first hint came in a Merck patent and publication which pointed out that ortho-biphenyl substituted compounds could preserve the activity.

The four statins that preceded atorvastatin.

The Merck compounds that inspired the pyrrole template used in atorvastatin.
Unsurprisingly, the Pfizer team asked whether the aromatic functionality described in the Merck compounds could be replaced by other ring systems. It is at this point that the magic of chance and serendipity enters the picture. Many homocyclic and heterocyclic moieties could be used in place of the o-biphenyl moiety, but Bruce Roth happened to have studied pyrrole chemistry extensively during his postdoc, so he decided to use pyrroles as a replacement. Roth himself admitted that at that point he decided to make pyrroles not because that was the rational decision but because it was what he was trained to do. Nor was the decision a straightforward one, as Jack Li reports in his excellent history of statins, “Triumph of the Heart”.

Pyrroles can be made easily using the Paal-Knorr synthesis discovered in 1884: the condensation of 1,4-diketones with amines. Paal-Knorr chemistry is versatile and is amenable to a variety of different substituted pyrroles (although as we will see later, it is sensitive to the steric environment of the pyrrole).
One of the initial questions asked was regarding the optimal spacing between the pyrrole and the mevalonolactone. Using a series of 30 or so analogs, the team found precise distance requirements between both the pyrrole and the mevalonolactone and the different substituents, especially at the 2 and 5 positions. A simple ethyl linker between the ring systems seemed to work best, A 4-fluorophenyl at the 5-position and an isopropyl at the 2-position also seemed to work best. The problem was that even this compound was only one-tenth as potent as a previous statin, mevastatin.
When medicinal chemists think they have extensively explored the SAR in a compound and still not seen satisfactory potency, the question is always whether it’s because they have missed some combination of substituents or whether it’s a fundamental liability of the basic scaffold. To understand this distinction, the team embarked on a molecular modeling exercise that, especially in this day and age, is remarkable in its simplicity. Since they already knew that the Merck compounds were active, they did a 2D overlay of the pyrroles with those compounds and found that there was a critical methyl group in one of the Merck aromatic rings that was occupying a space that was vacant in their compounds.
Overlay of the Merck compound with the pyrroles.
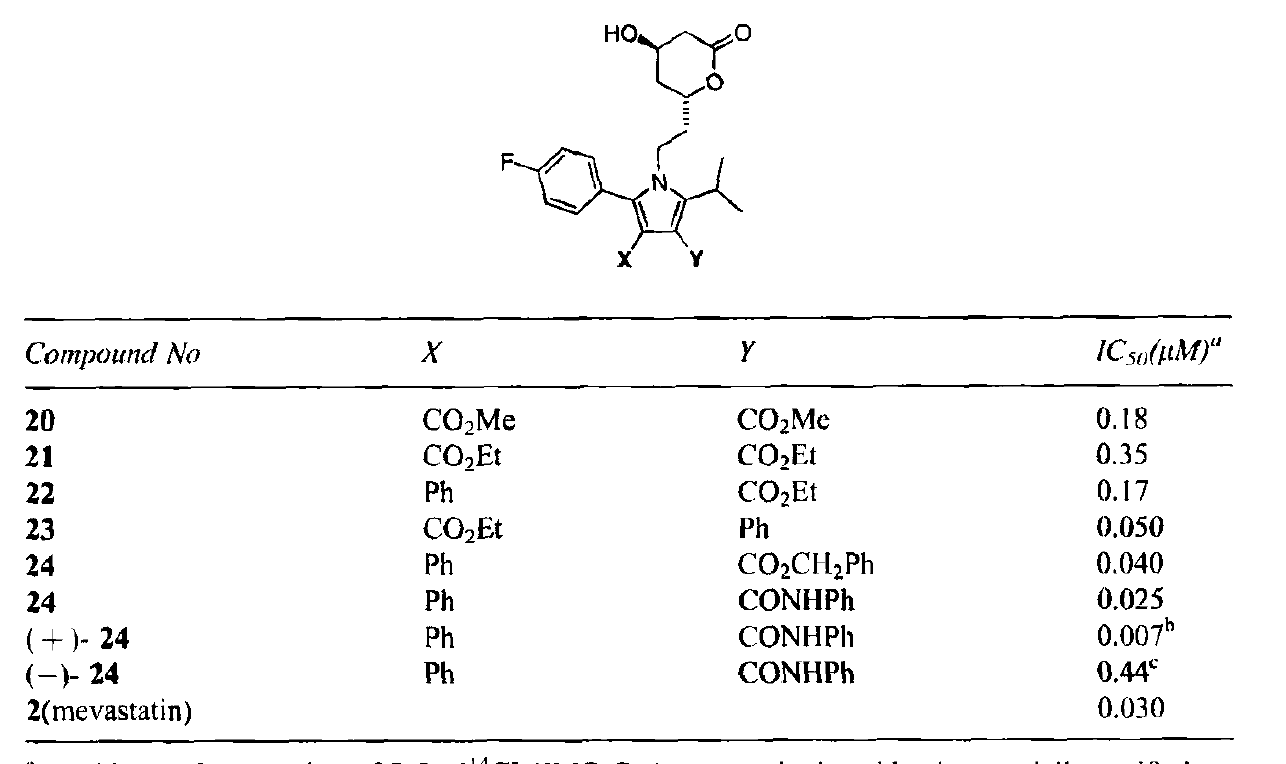
It was evident that substituting the 3 and 4 positions of the pyrrole series could enable the team to fill that vacant space. With this in mind, a variety of halogens were introduced at those positions for synthetic simplicity. It was found that the 3,4-dibromo analog had potencies similar to the earlier statins (IC50 28 nM).
Unfortunately, the potent dibromo analogs were found to display considerable toxicity in rodent models. However as found by others, the tox seemed specific to rodents and was also observed at high doses that would not translate to humans. This effect was observed most prominently with inhibitors with very high bioavailability, leading to excessive plasma and tissue concentrations (a cautionary tale that tells you that extremely high bioavailability may not always be good!).
Nonetheless, the team was faced with an important decision at this point since it was not clear whether the toxicity was substituent-specific or series-specific. They decided to approach the problem in two ways: first, by synthesizing non-halogen analogs at the 3 and 4 positions and by looking at alternative series. It was at this point that the limitations of Paal-Knorr chemistry became clear, since pentasubstituted pyrroles weren’t amenable to the chemistry, probably because of steric constraints imposed during the cyclization. To circumvent this issue, the team turned to the Huisgen [3+2] cycloaddition as an alternative. Considerable control over the regiochemistry could be obtained by picking the right amide and amino acid precursors. Yields were best with two electron withdrawing groups but were also satisfactory with a single electron withdrawing group.

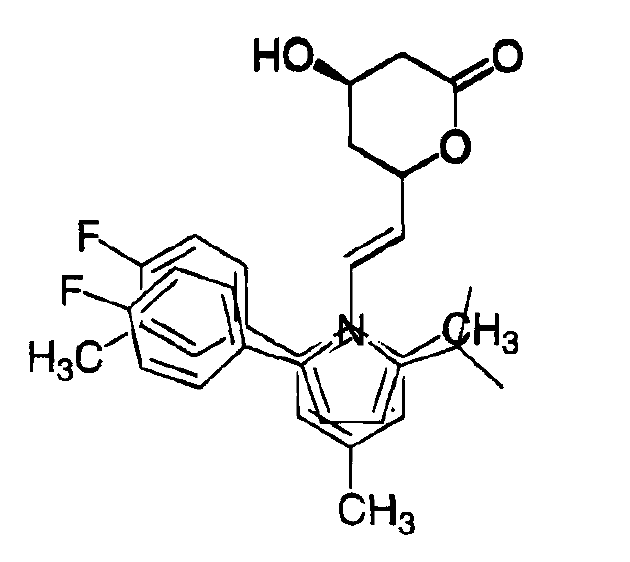
As can be seen, an analog with a Ph in the 4-position and a CONHPh in the 3-position was the most potent.
To investigate the high activity of this analog, the two enantiomers were separated. The R,R-stereoisomer turned out to be extremely potent (IC50 7 nM). Initial testing revealed that this analog had potency and efficacy equivalent to lovastatin.
Having identified a satisfactory pyrrole analog that was competitive with previous statins, the question before the team now was whether to go with the racemate or the pure stereoisomer. In its development, Sandoz had picked the racemate, but there were obvious reasons for going with the pure isomer (a strategy adopted by many companies): halving the material necessary and having a superior product in the marketplace that would compete with other successful products 10 years after their discovery. One of the critical decisions made at this juncture was to bifurcate the synthetic route: one at Discovery Chemistry in Ann Arbor, MI and the other at Chemical Development in Holland, MI.
There was an enormous amount of chemistry done by both groups to develop a scaled-up route to the pure enantiomer. The Ann Arbor group attempted a Paal-Knorr synthesis to make the tetrasubstituted pyrrole (recall that the pentasubstituted version precluded this chemistry) and introduce the CONHPh group later. Creative efforts were also made to install the 5-R-hydroxyl on the mevalonolactone ring. While these efforts were successful on a small scale, the linear nature of the synthesis and the low-temperature nature of the reactions involved did not lend themselves to scale-up. So the Holland group was forced to develop an entirely different approach which busted the myth of Paal-Knorr chemistry not working on pentasubstituted pyrroles (the secret turned out to be the use of a full equivalent of pivalic acid)!

By the time these extensive studies had been done by late 1989 and the team was ready to file an IND, three statins had already been approved by the FDA. This must have been disheartening to the team, but fortunately by this time, preclinical studies were showing a superior profile for atorvastatin compared to the others in lowering both total and LDL cholesterol. This positive data, along with a scaled-up synthesis of the compound, encouraged Pfizer leadership to move atorvastatin into clinical trials. The results were very impressive; a high dose of the drug reduced LDL cholesterol by up to 60%. The rest is history.
I love the atorvastatin story for several reasons: it shows the value of simple patent analysis and primitive molecular modeling; of being stubborn in persisting with a scaffold even against skepticism; of creative, dogged, collaborative synthetic efforts to beat a recalcitrant problem into submission; of close-knit, parallel interactions between discovery and development; and of courageous leadership that makes crucial decisions to halt or progress particular projects. Today, millions of patients can owe their health - and their lives - to the work done by Bruce Roth and the other scientists at Pfizer.
References:
Roth, B. Prog. Med. Chem., 40, 1, 2002
Lie, J. J.; Triumph of the Heart: The Story of Statins, 2009